

数据自身的质量在很大程度上决定了分析结果的准确和可靠,随着Hi-C技术在三维基因组学上的快速推广,对于Hi-C数据本身的质量和测序深度也逐渐引起研究人员的重视。同时对该技术的进一步优化和改进使之能够在更少的细胞起始量及测序量达到更高分辨率也成为了一个技术发展新的热点。本文旨在对Hi-C及相关技术的发展进行简略地介绍,并对Hi-C数据展示的无效数据进行分析,以期能让读者能更清晰地理解无效数据的组成,在后续的实验过程中能更好地改进实验方法,获得一份可靠的Hi-C的数据。
2009年Erez Lieberman-Aiden在3C的基础上,独创地在粘性末端添加了生物素,使得嵌合片段能被链亲和素特异性富集,发明了第一代 dilution HiC技术1)。Hi-C的发明与二代测序完美结合,解决了5C在全基因组水平构象数据量瓶颈的问题,使得在全局范围内研究三维结构成为可能。
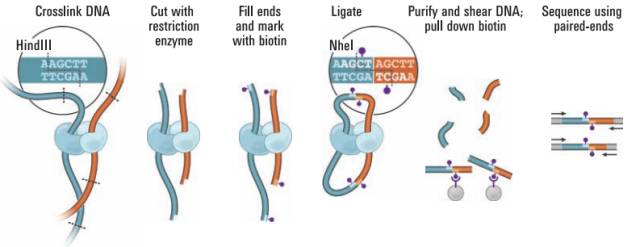
图1.Hi-C实验原理
早期的实验方案认为,去垢剂SDS在对交联的细胞核进行处理时,即使是低浓度的SDS(0.3%-1%SDS)在加热到65℃时,会导致细胞核碎裂,基因组的DNA会释放到溶液中,因此第一版本的Hi-C,在酶连反应体系下,选用了近8ml的大连接体系。后来,4C研发人员通过用显微镜观测SDS处理细胞核,发现细胞核仍然维持在一个较为稳定的核结构。在共聚焦显微镜下观察,1%SDS处理细胞核,会导致细胞核的通透性发生改变,但很少细胞核发生裂解。
另一个影响交联反应的因素是温度,通常认为65℃以上,在有NaCl存在的情况下,甲醛交联的DNA会发生解交联现象,从而影响染色质构象的稳定。
在第一版本的Hi-C选用了65℃ 1%SDS处理细胞核10min,从最终的数据看染色质间的互作数据高达27.1%-65.3%。通常认为染色质是独立折叠定位在细胞核中形成染色质领域的,因此染色质间的数据通常会认为是无效数据(bais)。
直到2012年Chen Lin实验室意识到细胞核的扰动会影响到染色质的高级构象,因此他们在Hi-C实验的基础上,将生物素标记在蛋白上,将反应体系固定在磁珠上,使得反应体系扰动更小,更稳定,TCC2)获得的数据结果表明该方法可以显著降低染色质间的数据占比例valid pairs的比例。
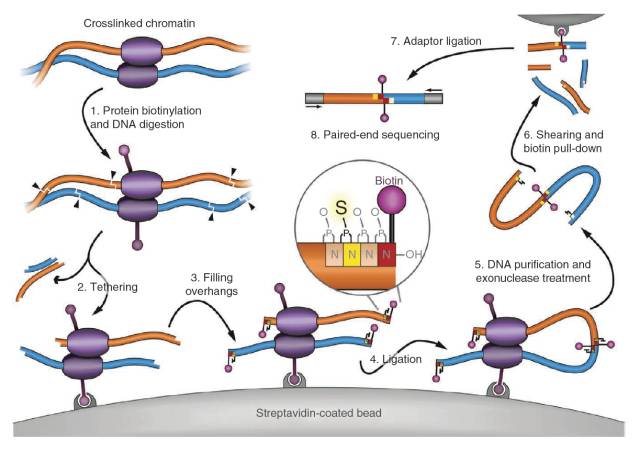
图2.TCC实验原理
2014年EreZ对Hi-C的实验进行了进一步的改进3),他们在SDS处理细胞核的步骤选用了更温和的0.5%SDS 62℃处理5-10min,而细胞连接的体系也降低到1ml,值得一提的是他们在文章中尝试了未交联的HiC实验。发现除了噪音增加外,得到了与正常HiC类似的热图。
In situ Hi-C的改进使得染色质间的互作数据进一步降低,实测数据显示Trans-interaction其占valid pair的比例在20%左右。
在2015年,又有研究将SDS的处理条件更换成37℃ 60min,他们认为该方法可以更大程度维持细胞核的稳定性,提高intra/inter数据的比例4)。
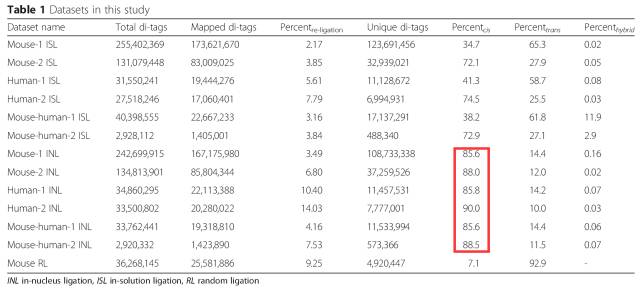
表1:in solution Hi-C与 in nucleus Hi-C数据比较
随着对HiC数据的进一步认识,研究人员发现一些超近距离的连接(<20Kb的数据)可能并不是有意义的由蛋白介导的空间上靠近的互作,而可能就是线性距离较近而引起的随机连接,因此引入了这一参数来评判数据的质量。
为了更好地去除随机连接导致的bais,有研究利用统计模型认为三片段的连接可减少随机连接的可能性,因此他们采用了类似于ChIA-PET
的方法,在连接反应过程中,添加一个带有生物素的bridge-linker5),通过富集带有linker的嵌合片段,来改善实验中存在的随机连接可能性。作者自测的结果表明,添加linker后染色质内的互作比例比in situ Hi-C和HiChIP都有显著改善。
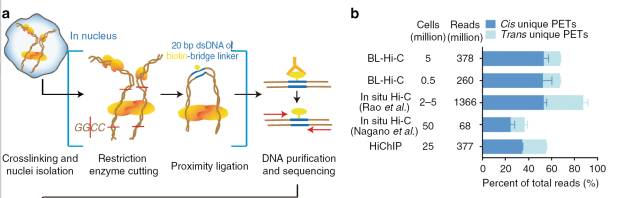
图3.BL-HiC实验原理图
除了cis/tran作为评判Hi-C数据的质量以外,Hi-C数据中还存在大量的无效数据,它们的存在会影响数据的有效利用率,以下篇幅将逐一进行介绍。
为了更好地理解Hi-C数据,在此我们简要介绍下基于illumina平台的二代测序文库。
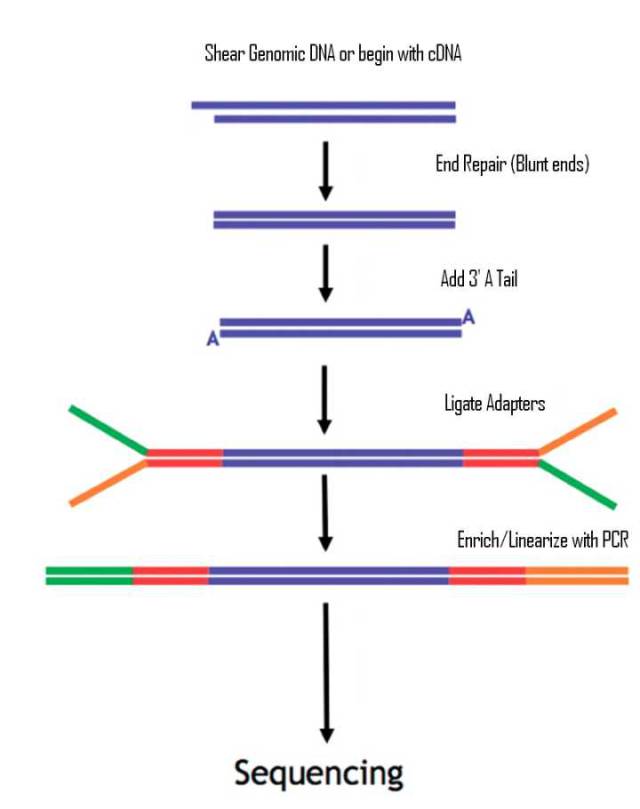
图4.二代测序文库建库示意图
在标准的二代文库中,DNA片段通过末端补平加A;再添加adapters ;此时reads的两侧各带发卡结构P5/P7的测序接头;为了获取足够上机的DNA文库,通常还需要进行一轮扩增;扩增后的文库两端各带一种测序接头。
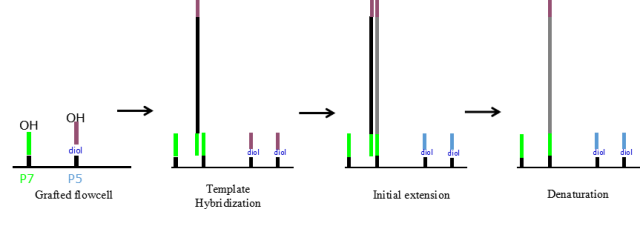
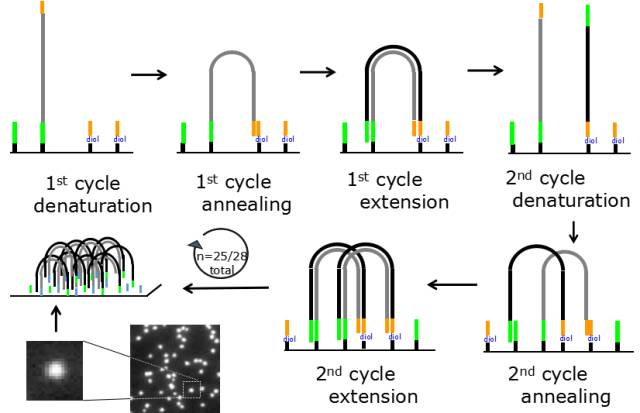
图5.桥式PCR
在pool DNA到芯片上时,文库片段首先anneal在芯片的测序接头上;然后用DNA聚合酶进行扩增,DNA生长在芯片上;经过25-28轮的扩增,每条reads被扩增至数以千计的拷贝,此时就可以利用添加可逆的终止子来检测碱基的组成。通过150轮添加可逆终止子并采集信号即可完成测序。
由于DNA聚合酶的自身的偏性,GC含量相对合适的片段及小片段更容易在芯片生长阶段得到富集。小片段(<150nt)在测序过程中,由于两端各读取150个碱基,就极可能将DNA插入片段读通,从而这部分的DNA就可能被检测到adapter污染。
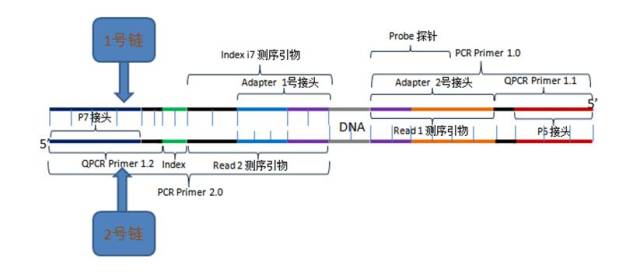
图6.测序read示意图
Hi-C标准文库是标准的Chimera结构,在将两端序列进行比对到基因组上时,理论上两侧pair ends可以分别比对到基因组的两个座位。由于DNA在碎片化过程中,剪切是随机的,因此酶切位点末端补平形成的junction fragment很可能分布在一侧的reads中,常规的比对分析是很难处理chimera的。在HiC-Pro6)和HiCUP7)软件中,他们会去识别理论的junction fragment。如HiC-Pro在比对时先进行Global Mapping,后将unmapping的reads用junction fragment序列进行识别并切割,再进行local mapping,最终将数据进行合并。
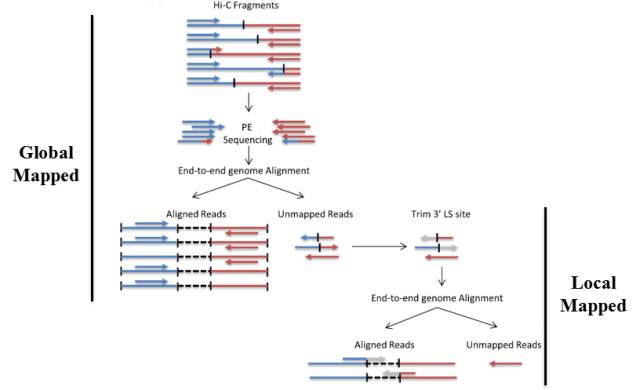
图7.HiC-Pro的两种比对策略
在实际比对中即使采用两步比对方式,仍有可能是只有一端序列能比对到基因组中,另外一端无法识别到基因组中,这种情况我们将其归类为Singleton。它产生的原因可能有①adapter污染(先前数据没进行过滤);②另一侧数据质量较差,多数为N的区域;③DNA片段被降解或酶切反应产生星号活性。同时片段过短,150碱基已经读通了生物素标记的位点,但是该位点不是正常的junction fragment。在植物样本中,singleton较为常见,可能与细胞壁破碎不完全,部分细胞质成分进入到反应体系影响酶切有关。
有些植物的基因组存在大量的重复序列,如玉米中85%的序列被认为是重复序列。这对要求两端都要唯一比对的HiC而言是巨大的挑战,一旦有一段比对到两个或两个以上的位点,该reads就将被归类到Multiple mapped reads中。
如果在比对过程中,global mapping 和 local mapping均无法将序列识别到特定的位点,这种序列会被归类到Unmapped reads。它可能产生的原因是基因组的组装完整度较差,基因组中存在大量的gap无法识别,被填充为NNNNN。另一个原因是酶切片段较碎,多个酶切片段连接在一起,无法识别到特定座位。
如果两侧数据都能比对到基因组的数据会被统一认为是Unique mapped reads,此时对于动物基因组,unique mapped reads 占测序量(clean reads)50%以上应是可接受的范围。对于植物样本,尤其是重复序列较多的样本,unique mapped reads 比例可能会急剧降低。
在获取unique mapped reads后,要进行进一步过滤,以识别真正有效的interaction reads。
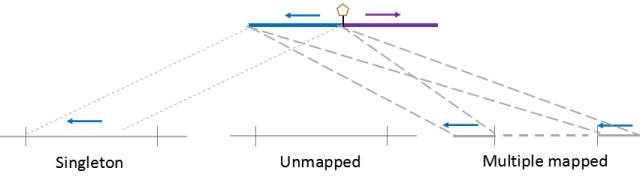
图8.三种比对过程识别的无效数据
根据HiC实验的基本原理PLA(proximity ligation assay):空间上相互靠近的片段更有机会被连接在一起。因此仅且仅有两个来源不同的片段连接在一起才会被认为是标准的文库片段。而这片段是指利用限制性内切酶酶切的Fragments,即唯有两个片段能分别比对到两个不同的酶切片段上,且实际片段大小(observe)符合理论的片段大小,在分析是才会将其归类到valid pairs中。
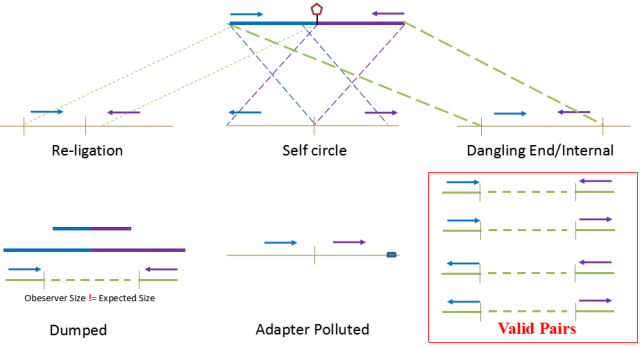
图9.Hi-C数据过滤
因此在分析过程中会将部分无效的数据进行过滤,首先是如果两个片段原本通过一个酶切位点连接在一起,在HiC文库中如果该片段即使酶切后添加生物素仍然连接在一起,该片段会被归类到Re-ligation reads中;
而如果两个的reads比对到同一个fragment,但是方向相反,则该reads会被认为是首尾相连形成了Self-circle ;
如果pair end 同时比对到一个酶切片段上,则该片段会被认为是Dangling ends;
如果有一个发现是adapter污染,该reads会被认为是Adapter polluted;
如果两侧的end均能比对到基因组的两个酶切片段中,但是观测到的片段大小与理论的片段大小不一致,则该片段会认为是错误连接而被归类为Dumped reads;
只有比对到两个酶切片段且片段的理论值等于实际值的reads,才会被认为是Valid pair reads。
在这里我们解释下Dangling ends和Dumped的成因。
Dangling ends 主要来源于两部分,①经DNA连接酶连接反应后,携带生物素的DNA片段末端并未形成嵌合片段,在末端生物素切割的(klenow)时又未将末端的生物素去除,从而进入到最终的文库中;②磁珠洗脱步骤未完全将非特异性结合的DNA洗脱下来。有文章报道,只有将Dangling Ends的比例控制子啊10-45%以下才会被认为是成功的Hi-C文库8)。
Dump的主要原因在于酶的星号活性导致切割位点不在经典的位点,这有可能是酶切时间过长或反应体系中盐离子浓度和种类不合适导致的;另外一个原因是片段被DNA外切酶降解,使得片段的大小发生了改变。
获得了interaction reads后,要去除文库中完全一样的reads,因为这部分可能是由于PCR扩增导致的Duplication,去除Duplication后,Valid pairs数据可用于后续的滑bin统计分析了。
最后,对分享的内容进行总结。
判断HiC的文库是否合格的一个重要的指标是cis/trans的比值,一般认为cis interaction比例越高,表明该数据的质量越好。如果tran interaction的比例高于cis interaction的比例,则要慎重检查实验操作步骤是否出现纰漏。
对于植物样本,尤其是大基因组的植物样本,其unique mapped的比例可能较低,此时为了达到足够的数据量,需要提高测序深度;然而如果对于人鼠等动物样本,如果unique mapped ratio较低则可能是实验原因。
在unqiue mapped数据过滤步骤中dangling ends 过高可能是末端生物素去除不完全或磁珠洗脱步骤中出现问题所致。如果dump的比例过高则可能是样品发生了降解或星号活性。
最后一步去除PCR duplication,如果该步骤中duplication比例过高,则表明PCR循环数过高导致。
Hi-C实验步骤繁多,一份好的Hi-C实验数据需要实验人员针对不同的样本进行实验优化及在整个实验周期每个步骤用心地操作。出现不如人意的实验结果对于新手而言是正常的,此时就要对数据进行仔细分析,并将自己融入到实验的每个细节中细细体会,才会有所收获。最后给大家一个建议,多看看最近发表的文章,比较每个protocol的细微差别,如颉伟老师9)和陈阳老师5)今年发表的文章。相信看完后,会有自己的体会。
参考文献
1. Lieberman-Aiden E, Van Berkum N L, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome[J]. Science, 2009, 326(5950): 289-293.
2. Kalhor R, Tjong H, Jayathilaka N, et al. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling[J]. Nature biotechnology, 2012, 30(1): 90-98.
3. Rao S S P, Huntley M H, Durand N C, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping[J]. Cell, 2014, 159(7): 1665-1680.
4. Nagano T, Várnai C, Schoenfelder S, et al. Comparison of Hi-C results using in-solution versus in-nucleus ligation[J]. Genome biology, 2015, 16(1): 175.
5. Liang Z, Li G, Wang Z, et al. BL-Hi-C is an efficient and sensitive approach for capturing structural and regulatory chromatin interactions[J]. Nature Communications, 2017, 8(1): 1622.
6. Servant N, Varoquaux N, Lajoie B R, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing[J].Genome biology, 2015, 16(1): 259.
7. Wingett S, Ewels P, Furlan-Magaril M, et al. HiCUP: pipeline for mapping and processing Hi-C data[J].F1000Research, 2015, 4.
8. Belton J M, McCord R P, Gibcus J H, et al. Hi–C: a comprehensive technique to capture the conformation of genomes[J]. Methods, 2012, 58(3): 268-276.
9. Ke Y, Xu Y, Chen X, et al. 3D chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis[J]. Cell, 2017, 170(2): 367-381. e20.
1F
dump pair 观测到的片段长度与理论片段长度不符合是什么意思??什么算观测到的片段长度,什么算是理论片段长度???