

日常工作的窘境
谈基因浏览器的必要性,不需要扯“各种基因组序列以及高通量测序数据爆炸性增长,满足基因组可视化、大规模基因组数据分析和应用需要”这些有的没的,只需要从日常实际需求出发就可以。
在日常数据处理和分析工作中,根据分析项目的不同我们会面对各种各样的文件,比如mapping得到的SAM或者BAM文件,在此基础上转化而来的记录各种(定量)信息的bedgraph文件、Wig文件或者tdf文件,亦或记录variant信息的VCF文件。以至于有一种比较调侃的说法,所谓“生物信息”就是整天和各种格式的文件打交道,转换来转换去。
那么,跑了pipeline以后拿到各种文件,如何形象方便地利用这些文件进行后续分析或者向合作者展示信息呢?
可能每个做生物信息分析的人电脑里都有一个软件IGV,它在高通量数据结果可视化领域非常强大,支持读取和展示各种类型的数据。IGV截图在各种文献里的出镜率也不要太高。

但类似于IGV这样的应用程序通常有两个硬伤让人比较崩溃。
- 程序响应速度慢:16G内存配置的台式机,但凡要加载个BAM文件、搜个具体基因和位置或者比较放肆地放大缩小,等上几分钟那都不是事儿。
- 结果不方便分享:有时候合作者想看十几个基因的表达情况,不得不发一堆截图过去,说不定对方什么时候又想看什么,还得让自己一直处于待机状态。
有没有一种方法既能提高数据的加载速度又能方便地与别人分享?
至此,基因浏览器就可以出场了。
基因浏览器是什么
所谓基因浏览器,首先你可以把它理解为一个网站(网页),只不过使用它不再需要从HTML和CSS写起,也不用JS来编写各种function。其次它的功能是用来展示各种基因信息,比如基因的外显子内含子,比如转座子和重复序列或者SNP等。
这样一来,好处之一是数据呈现的压力从本机转移到了后端服务器;好处之二是其本身的网站属性让你我都可以随时访问查看。
常见的基因浏览器
基于Web技术的基因浏览器目前有很多,按道理应该先罗列一些常用基因浏览器的优缺点,再引出要推荐的主角。但是篇幅有限,只提两个。
Map Viewer 是NCBI提供的一个可视化分析工具,通过Map Viewer可以了解感兴趣的基因在基因组中的位置、基因序列、内含子和外显子排列等信息。但它可视化界面不友好,操作上不支持鼠标拖拽选择区域,更重要的是NCBI 团队对于Map Viewer已经不再开发了。关于Map Viewer 可以查看链接 https://www.ncbi.nlm.nih.gov/books/NBK21089/
**UCSC Genome Browser **是目前知名度最高的基因浏览器之一,这个网站提供了80多个物种的各种基因组信息。但用的Web技术相对落后,性能的制约会对用户的访问频率进行各种限制,每拖动一次都要看一会儿进度条。相关内容可以查看链接 http://gmod.org/wiki/GBrowse
JBrowse 是今天要介绍的主角。它是GMOD开源项目的一部分,想了解这个开源项目可以查看它的官方网站http://gmod.org/wiki/Main_Page
下面是对GMOD的一句话介绍:
Generic Model Organism Database project, a collection of open source software tools for managing, visualising, storing, and disseminating genetic and genomic data
在GMOD的众多项目中最出名的应该是Galaxy,而JBrowse则是GMOD之前一款基因浏览器GBrowse的继任者。

JBrowse 基因浏览器特点
首先来看一下官方主页的自我介绍,说的已经很清楚了。
JBrowse is a genome browser with a fully dynamic AJAX interface, being developed as the eventual successor to GBrowse. It is very fast and scales well to large datasets. JBrowse is javascript-based and does almost all of its work directly in the user’s web browser, with minimal requirements for the server.
完全基于HTML5和Javascript构建的JBrowse通过AJAX技术实现了数据的异步加载,所以响应速度非常快。由于Javascript将大量的计算工作在前端完成,服务器端只需要向浏览器客户端发送静态文件,因此也极大程度减轻了服务器端的负担。
- Fast, smooth scrolling and zooming. Explore your genome with unparalleled speed.
- Scales easily to multi-gigabase genomes and deep-coverage sequencing.
- Supports GFF3, BED, FASTA, Wiggle, BigWig, BAM, VCF (with tabix), REST, and more. BAM, > > - BigWig, and VCF data are displayed directly from the compressed binary file with no conversion needed.
- Very light server resource requirements. In fact, JBrowse has no back-end server code, just tools for formatting data files to be read directly over HTTP.
在JBrowse中可以随心所欲的平滑动态移动和缩放。支持多种格式的数据,比如GFF3, BED, FASTA, Wiggle, BigWig, BAM, VCF (with tabix)。其中Bigwig和bam这类二进制文件不需要转换就可以直接展示。
另外,JBrowse经过几个版本迭代升级,现在已经非常容易上手。按照官方提供的Quick-Start Tutorial,前后只需要十几分钟时间就可以完成基本配置呈现出想要展示的数据。
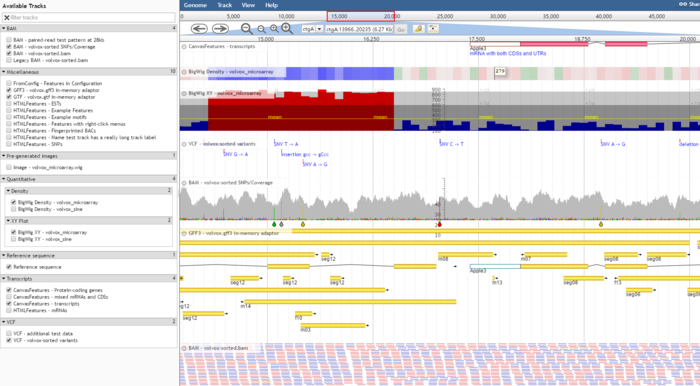
JBrowse 安装
在JBrowse官网下载压缩包,最新的版本一共也就只有4.88 MB大小。
为了后续JBrowse可以通过web服务器访问(毕竟它是一个网站),首先在/var/www/下给它准备一个目录,比如jbrowse。配置好之后,就可以通过 http://.*..*/jbrowse 来访问了。
# 建立有写权限的目录 sudo mkdir /var/www/jbrowse; sudo chown `whoami` /var/www/jbrowse; cd /var/www/jbrowse; # 下载最新的安装包 curl -O http://jbrowse.org/releases/JBrowse-1.12.1.zip # 解压之后进入目录 unzip JBrowse-x.x.x.zip cd JBrowse-x.x.x #运行目录内的setup.sh文件 ./setup.sh
setup.sh运行后,会自动安装一些需要的perl环境并且会自动生成一个example。正常的话,setup.sh运行之后就可以使用JBrowse了,如果在安装的过程中出现了错误,可以去setup.log文件查看有什么报错信息。
JBrowse 配置
总体说明
因为JBrowse实质是一个网站,也就意味着可以定制页面上绝大多数展示的内容,比如导航栏的样式或者颜色等。因为它完全基于Javascript开发,也意味着在JBrowse页面上进行的绝大多数交互动作都可以利用各种回调函数来进行定制,比如右键单击基因显示什么菜单栏。
JBrowse 主目录下的CSS目录可以供你对网页的CSS属性进行更改;主目录下的jbrowse.conf和jbrowse_conf.json文件可以起到全局配置的作用;主目录下的docs目录则包含了各种说明文档。
如果具有一定的前端开发或者JS基础,完全可以用它实现各种各样的需求,当然前提是有没有这个需要。下文仅仅列举一些基本的配置和注意事项,更深入的内容我也在学习和尝试。如果感兴趣或者有需求的话可以参考官方详细的配置说明。
JBrowse支持两种配置文件格式,一种是JSON格式(文件后缀为.json),另一种是文本格式(文件后缀为.conf)。
JSON是一种轻量级的文本数据交换格式,和XML相比更小更容易解析,它无需解析器就可以通过JS的内建函数生成对象。尤其适合AJAX进行传输,JBrowse支持它几乎是必然的。而文本格式少了JSON格式看似不友好的花括号和引号,更具有可读性。
JSON格式示例(来自官网)
{
"tracks": [
{
"urlTemplate" : "volvox-sorted.bam",
"storeClass" : "JBrowse/Store/SeqFeature/BAM",
"type" : "JBrowse/View/Track/Alignments2",
"label" : "BAM_track",
"key" : "My BAM track"
"style": { "color": "red" }
}
]
}文本格式(来自官网)
# BAM track with a new callback
[tracks.mytrack]
storeClass = JBrowse/Store/SeqFeature/BAM
type = JBrowse/View/Track/Alignments2
urlTemplate = myfile.bam
key = My BAM track
style.color = function(feature) {
/* comment */
return 'red';
}如果使用文本格式,有两点需要注意
- 中括号"[]"内"tracks.“后面的名字应该是唯一的,而且track的名字不应该在有”."符号了。
- 使用回调函数时,所有的回调内容,也就是return那里的内容都不可以在最左边,结束的右半部份花括号也不可以。
准备参考序列
安装成功之后首先要做的是格式化参考序列,支持的格式有fasta, gff,或者是用samtools faidx处理过的indexed fasta 文件。参考序列生成的track 会为后续所有文件提供一个坐标,一直放大后参考序列的碱基也会显示出来。
通常我们使用某一个物种的genome fasta 文件,默认情况下,每一条染色体会独立为一条参考序列。如果想把RNA或者蛋白序列当成参考序列也没有问题。
需要注意的是所有数据默认都会输出在out目录中,如果你想用JBrowse展示不同物种的信息,最好使用--out参数指定单独的目录。
在这里提前说明,后面所有基于命令行设置的配置信息都会自动生成在tarckList.json文件中。
准备参考序列需要用到的是bin目录下的prepare-refseqs.pl脚本
bin/prepare-refseqs.pl --fasta yourpath/genome.fa
准备特征数据
所谓特征数据(feature data)就是指基于位置的注释信息,比如常见的gff文件和bed文件。如果想把gff文件和bed文件导入生成feature track 需要用到bin目录下的flatfile-to-json.pl
基本的命令比较简单
bin/flatfile-to-json.pl --[gff|gbk|bed] <flat file> --tracklabel <track name> [options]
只有两个必要的参数,--gff/bed用来指定数据格式,而--tracklabel 用来给track设定ID(track 的身份证号),非必需的--key参数可以理解为track 的名字。
在这个命令的众多参数中有一个要提一下--trackType ,它的两个选项分别是CanvasFeatures和 HTMLFeatures。
这里又涉及到了前端的一点概念。
所谓canvas 是HTML5中引入的新元素,用来定义图形,而这个图形通常需要通过Javascript的API进行绘制。通过JS可以画出各种形状和渐变的颜色。它的优势之一是非常适合进行动态渲染以及输出实时数据。
针对CanvasFeatures和 HTMLFeatures,JBrowse分别提供了两套完整的配置方案,具体内容不展开介绍了。只是展示一下两者之前不同的效果。
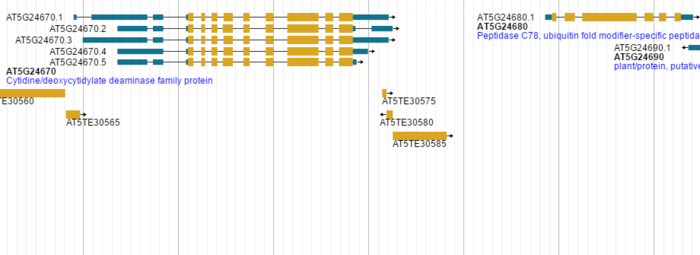

针对feature track 有更多的高级功能可以进行定制。
比如鼠标左击显示什么内容或者鼠标滑过显示的内容(其实都是JS里的功能),进而可以和其它数据库网站联通(比如点击基因进入NCBI对应数据库);比如利用回掉函数,让一部分基因显示红色,让另一部分基因显示蓝色。
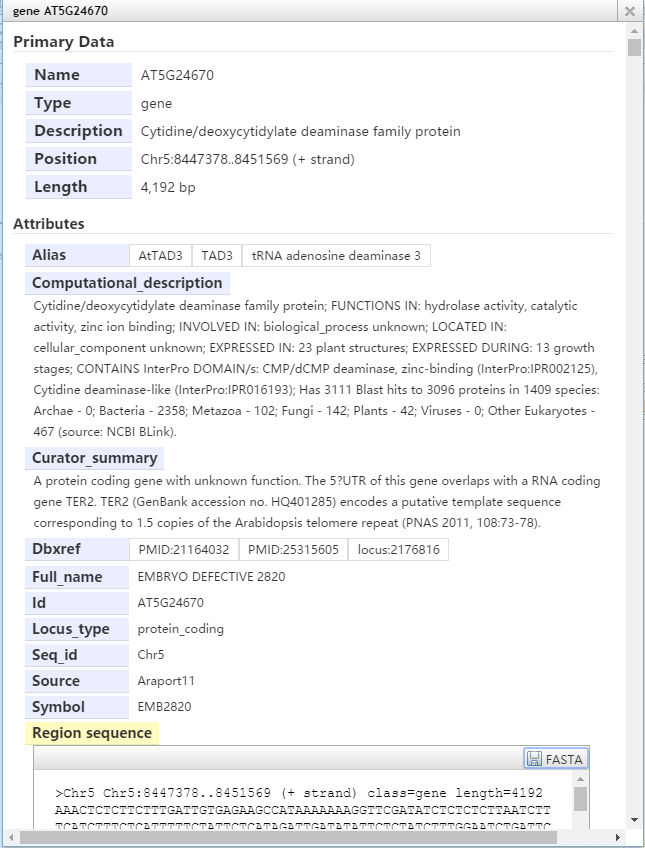
默认情况下,左击一个基因,可以显示它的名字类型和位置及长度,也可以显示该基因所有外显子和内含子的信息。
展示比对数据(BAM)
JBrowse支持导入BAM文件,但是前提是进行sort。
Alignments2 格式的track可以在碱基水平上展示错配,插入和缺失等信息。如果paired read 的mates缺失则会用红色强调。
如果想用这种格式来展示数据,需要的配置文件信息也非常简单,只需要写以下三行信息,其中urlTemplate 指文件存放的位置,指得注意的是,这里的路径为相对于配置文件而言所在的位置。
更多具体配置信息可以参考手册里的内容。
{
"label": "alignments",
"urlTemplate": "../sorted.bam",
"type": "JBrowse/View/Track/Alignments2"
"storeClass" : "JBrowse/Store/SeqFeature/BAM"
}另外一种格式叫做Alignments ,它展示的效果和IGV非常类似了,而且也支持各种回调函数和点击操作等。但如果文件过大,依旧可能出现加载较慢的问题。只需要在type中进行设置即可。

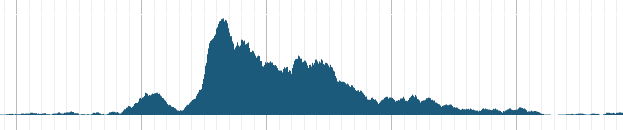
展示定量数据(BigWig)
BigWig文件是Wig文件的二进制压缩格式,可以通过bam文件再加上基因组信息转换生成。JBrowse 可以直接对BigWig文件进行展示无需转换。
对于BigWig文件有两种展示方式任你选择。
- XY-plot track
{
"label" : "rnaseq",
"key" : "RNA-Seq Coverage",
"storeClass" : "JBrowse/Store/SeqFeature/BigWig",
"urlTemplate" : "../tests/data/SL2.40_all_rna_seq.v1.bigwig",
"type" : "JBrowse/View/Track/Wiggle/XYPlot",
"outscale" : "local
"style": {
"pos_color" : "#FFA600",
"height" : 100
}
}以上是一个最基本的BigWig配置文件,在这里进行简单的说明。
label和key的含义和区别在上文已经提到过,而storeClass参数则指明文件格式,type参数指明使用XYplot展示数据。
而outscale设置为local是我比较常用的一个参数,它保证在浏览器显示界面内,根据已经显示的数据来自动就行调整,保证展示的效果。当然,也可以设置为global,通过整体的情况进行调整。如果不使用outscale参数,亦可以通过设置min_score和max_score来限定展示数据值的范围。
通过设置pos_color可以调整track的颜色,既然有pos,肯定就用neg_color参数。
另外,JBrowse会根据数据值自动调整track Y轴得刻度,默认显示在track中间,如果觉得碍事也可以放在左边或者右边。使用yScalePosition参数即可。
- Density track
基本的Density track 配置如下,不再过多解释了。
{
"label" : "rnaseq",
"key" : "RNA-Seq Coverage",
"storeClass" : "JBrowse/Store/SeqFeature/BigWig",
"urlTemplate" : "my_methylation_data.bw",
"type" : "JBrowse/View/Track/Wiggle/Density",
"bicolor_pivot" : "mean",
"style": {
"pos_color": "purple",
"neg_color": "green"
}
}
对于VCF文件,需要提前做的准备就是使用bgzip压缩之后再利用tabix -p vcf 建立index。
展示方式和BigWig类似,其中type要定义为 “JBrowse/View/Track/HTMLVariants”;而storeClass定义为"JBrowse/Store/SeqFeature/VCFTabix" 即可。
建立索引方便搜索
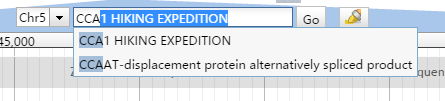
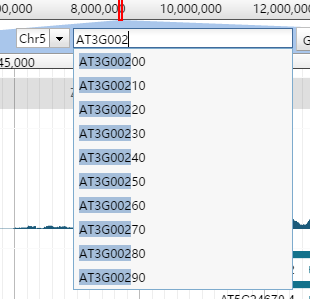
JBrowse界面有一个小方框会实时显示目前所展示的片段相对于整个参考序列的位置。
格式为Chr1:1652..1685 (35 b),其中冒号分割染色体和位置,而起始位置和终止位置用…分割,括号内显示长度。
但这个小方框绝对没有这么简单,后面的GO按钮其实已经暗示了它的搜索框属性。不过要想使用这个搜索功能还需要做一点准备工作。
JBrowse支持按照名字进行搜索和自动补全功能。只要对参考序列或者features建立了索引,就可以通过名字快速找到它们。当你通过 prepare-refseqs.pl和flatfile-to-json.pl生成了相应track后,只需要运行bin目录下的generate-names.pl脚本即可。
generate-names.pl原则上不需要任何参数,这个所谓的原则是指你的所有track都生成在默认的data目录中,如果不是就需要利用--out来指定track位置。如果你只想让某个gff或者bed文件可以被索引到,还需要通过 --tracks参数来指定需要生成索引的文件。通过--completionLimit参数,可以限定自动补全的字母个数,如果设为0意味着禁用这一功能。
不常用又很有用的功能
使用URL查询语句控制JBrowse
JBrowse 遵循基本的URL查询格式。所谓URL,中文全称是统一资源定位符,就是我们叫做网址的那一串字母。
JBrowse的URL如下所示:
http://<server>/<path to jbrowse>?loc=<location string>&tracks=<tracks to show>
- 其中“http”是一种传输协议,与之类似的还有https,ftp等等
- http后面紧接着的是服务器(域名或者IP地址)
- 再往后是路径,目录名之间用“/”分隔
- “?” 开始表示查询内容,也就是通过GET模式获得的参数,不同参数之间用“&”分隔,“=”连接参数名和参数值
有时候会发现有些网页的网址有很“%”后面还跟着一个数字一个字母,这是因为URL只支持ASCII字符集,如果你的URL包含了字符集之外的内容,URL则会对其进行转换。转换规则正是用“%”跟两位十六进制数来替换非ASCII字符。
具体到JBrowse 的URL。如果你选择查看多个track,不同的track之前会用“,”分隔,但是在浏览器中看的实际内容是tracks=DNA%2Catrack%2Cbtrack;LOC参数的传输格式是"Chromosome"+":"+ start point + “…” + end point,但是你看到的实际内容是Chr7:17566672…17572268。之所以如此,正是因为URL对“,”和“:”进行了字符的转换。
需要强调的一点是,JBrowse默认的data参数就是data目录,如果你的数据不在data目录下,“?”后首先给出的参数是“data=XXX”,然后才是“LOC”。另外,JBrowse还支持例如addFeatures和highlight等很多其它参数。
懂了这些,就有一个非常实际的应用场景。如果用户从我的A数据查询到了B基因后希望点击B基因可以跳转到JBrowse,我只需要把基因名字作为变量传到URL中,然后加一个<a><a/>就好。
track 分面搜索选择器
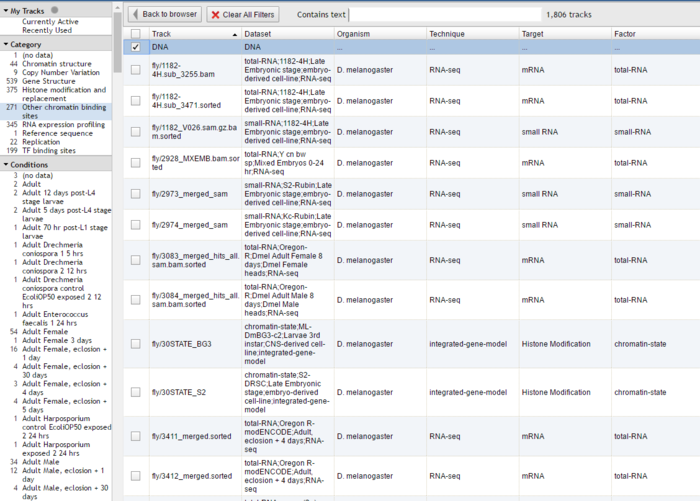
如果只用JBrowse来展示几个track自己没事的时候看看还好。如果实验室最近处理了500个Chip-seq数据,全部都显示在主界面的侧边栏就是一件很恐怖的事情。
为了解决这个问题,JBrowse提供了分面筛选的功能。
所谓“分面”(faceted)指的是事物几个不同的维度,比如在网上买一样东西,你可以从价格,性能和品牌几个不同的维度进行搜索从而更快地找到自己想要的东西。
针对大量track而言,可以先用csv格式来保存所有数据的元信息,比如第一列是Track名,第二列是数据来源,第三列是作者,第四列是数据类型。然后通过相应配置生成一个slector界面,允许用户通过对来源/作者/类型多个维度同时进行查询后展示其感兴趣的track。
设置 loading 页面
如果我们的数据因为需要而不得不展示很多bam文件,在使用的过程中难免会出现等待加载的情况。这个时候通过相应设置可以像普通网站那样生成一个loading页面,提醒用户“不要走开,精彩稍后继续”。
利用 PhantomJS 工具保存高分辨路截图
在JBrowse中看到了一个非常中意的展示效果想要保留或者用于后续文章的发表,JBrowse本身并没有提供这样的工具,但是结合 PhantomJS 可以轻松的完成。关于PhantomJS可以参考官网:http://phantomjs.org/screen-capture.html
JBrowse 网页端操作
上面提到的内容都是在服务器端完成的配置,其实在JBrowse网页端,也可以进行很多操作。
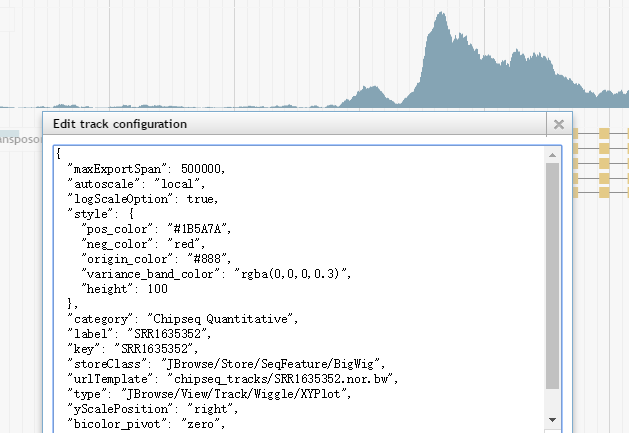
比如你可以添加本地的genome或者track文件,可以全局设置所有track的高度或者设置高亮区域。右键单击某个track的标签,然后选择“Edit config”,还可以在弹出的窗口中直接编辑json格式的配置文件。
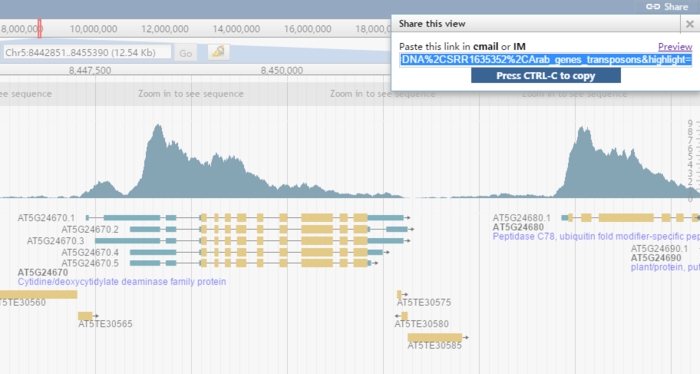
在页面右上角有一个share按钮,点击后会生成属于当前页面的连接,想分享给谁就分享给谁。
如果没有linux怎么办
写到最后,还有一个问题没有解决。
如果我的电脑是Windows系统,也根本没有服务器,甚至都没有一个浏览器,还能不能用JBrowse呢?
1.21.1版本的JBrowse 目前提供了的32位的windows桌面版和64位的OSX桌面版。如果你想体验一把docker的神奇,不妨试试这个 https://hub.docker.com/r/jbrowse/gmod-jbrowse/