

从这一节开始详细讲述正式流程的搭建,我将结合具体的例子努力争取将这个系列写成比GATK最佳实践更加具体、更具有实践价值的入门指南。整个完整的流程分为以下6部分:
- 原始测序数据的质控
- read比对,排序和去除重复序列
- Indel区域重(“重新”的“重”)比对
- 碱基质量值重校正
- 变异检测
- 变异结果质控和过滤
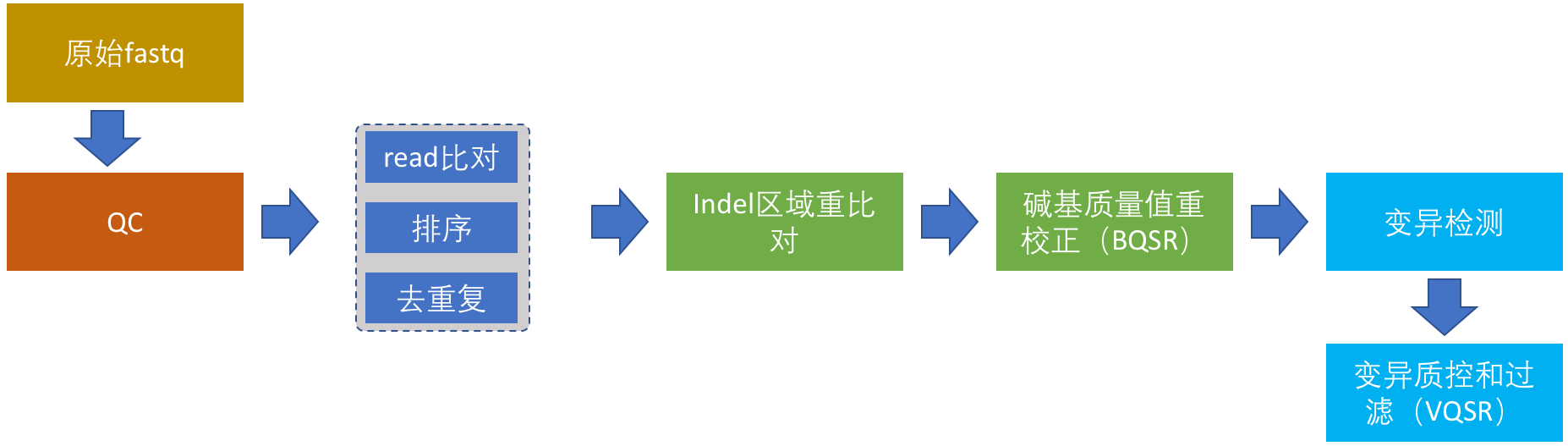
在这个图中,我把WGS数据分析流程的各个步骤和关系都画下来了。这个流程虽然只针对于人,但对于其它二倍体生物来说,同样具有借鉴价值。这6个步骤,接下来我也会进行详细介绍,在本篇文章中我们首先介绍原始测序数据的质控。
认识测序数据——数据质控的意义
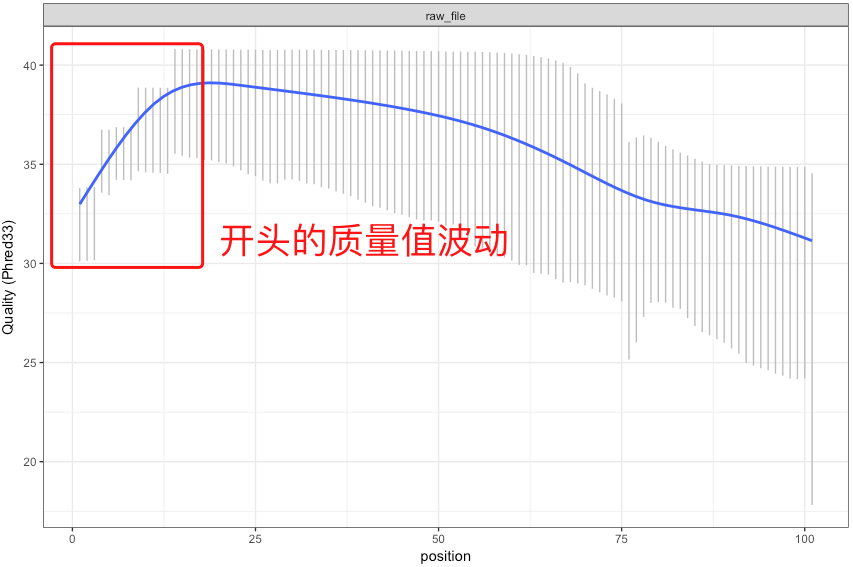
在第1节测序技术中,我们已经知道现在的NGS测序,以illumina为首基本都是运用边合成边测序的技术。碱基的合成依靠的是化学反应,这使得碱基链可以不断地从5’端一直往3’端合成并延伸下去。但在这个合成的过程中随着合成链的增长,DNA聚合酶的效率会不断下降,特异性也开始变差,这就会带来一个问题——越到后面碱基合成的错误率就会越高【注】,这也是为何当前NGS测序读长普遍偏短的一个原因。
【注】:有时候测序仪在刚开始进行合成反应的时候也会由于反应还不够稳定,同样会带来质量值的波动,不过这个波动一般都在高质量值区域(如下图)。
测序数据的质量好坏会影响我们的下游分析。但不同的测序平台其测序错误率的图谱都是有差别的。因此,非常建议在我们分析测序数据之前先搞清楚如下两个地方:
- 原始数据是通过哪种测序平台产生的,它们的错误率分布是怎么样的,是否有一定的偏向性和局限性,是否会显著受GC含量的影响等;
- 评估它们有可能影响哪些方面的分析;
第一点是我们认识数据质量的第一步,也是我们一定要去知道的地方。除了看官方的资料之外,最好的做法是自己分析。
虽然随着NGS测序数据变得越来越普遍,整体的测序质量和错误率分布情况大家也都了解一些。在实际的工作中,我也常常发现很多人其实并不十分关心这个数据到底长啥样,拿到之后,就直接跑过滤流程,也不管这些参数或者工具是否真的是合适的,更加不看看过滤后的数据和过滤前到底有什么不同。尽管,大多数情况下问题不大,但是我想跟大家说的是: 认识你的数据,不要相信你的工具! 这样也能够更好地避开很多不必要的坑。
因此,在本文中我将谈谈该如何更好地去认识一个测序数据,而不只是简单地告诉大家一个质控流程,当然,这也是本篇文章将要进行介绍的内容。至于第二点其实需要视情况而定,例如你的测序深度是多少,检测变异的时候,变异的位点是否过于集中在read的末尾,比对的时候是否会出现了一定的正反链偏向性等诸如此类的问题;又或者我们在进行基因组序列组装的时候,由于对read的中出错的碱基更加敏感,因此往往需要进行更严格的切除,不然会由于这些错误的碱基消耗更大的计算资源和时间。
那么说大地该如何认识一个原始的测序数据(fastq data)呢?一般我们可以从如下几个方面来分析:
- read各个位置的碱基质量值分布
- 碱基的总体质量值分布
- read各个位置上碱基分布比例,目的是为了分析碱基的分离程度
- GC含量分布
- read各位置的N含量
- read是否还包含测序的接头序列
- read重复率,这个是实验的扩增过程所引入的
以上,这几个地方都弄明白了,那么这个数据的基本情况也就差不多都清楚了。
我们首先来说说read各位置的碱基质量分布。在第2节里面,我们已经知道该如何通过简单的Python代码计算出read的质量值和碱基的测序错误率了。但对于成千上万的read来说,这样做并不合适,我们需要更直观的表达方式——画出来,正所谓 一图胜千言!目前也有很多现成的工具可以高效地来完成这样的事情,比如用得最广的FastQC,它是一个java程序,能够用于给出测序数据的QC报告,报告中会同时给出上述几个方面的数据图,并提示原来的数据可能还存在着哪些问题。它可以很好地帮助我们理解测序数据的质量情况,但缺点就是 图!太!丑!
在做read质量值分析的时候,FastQC并不单独查看具体某一条read中碱基的质量值,而是将Fastq文件中所有的read数据都综合起来一起分析。下图是一个测序质量非常好的read各位置碱基质量分布图(如下图)。
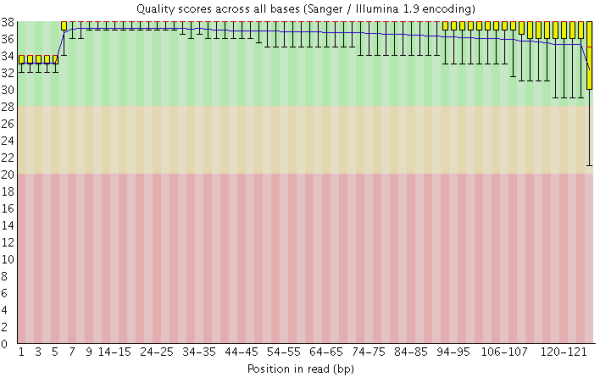
这个图的横轴是read上碱基的位置,纵轴是碱基质量值。在这个例子中,read的长度是126bp(来自HiSeq X10的测序结果),这应该算是比较长的二代测序序列了。我们可以看到read上的每一个位置都有一个黄色的箱型图表示在该位置上所有碱基的质量分布情况。除了最后一个碱基之外,其他的碱基质量值都基本都在大于30,而且波动很小,说明质量很稳定,这其实是一个非常高质量的结果。而且我们可以看到图中质量值的分布都在绿色背景(代表高质量)的区域。
那如果是质量很差的结果看起来会是怎么样的呢?我手边一时找不到这样的数据,就在网上找到了一个代替品,样子如下:
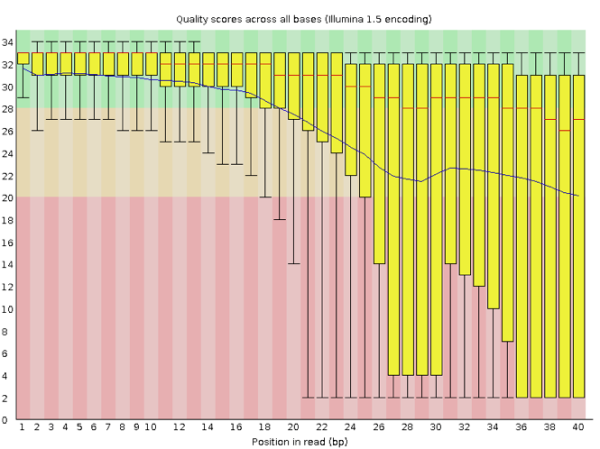
在这个图中我们可以明显看到,read各个位置上的碱基质量分布波动都比较大,特别从第18个碱基往后全部出现了大幅度的波动,而且很多read的碱基质量值都掉到非常低(红色)的区域中了,说明这个数据的测序结果真的非常差,有着大量不及格的read。最好的情况是重新测序,但如果不得不使用这个数据,就要把这些低质量的数据全都去除掉才行,同时还需留意是否还存在其他的问题,但不管如何都一定会丢掉很大一部分的数据。
除了上面read各位置的碱基质量值分布之外,FastQC还会为我们计算其他几个非常有价值的统计结果,包括:
1)碱基总体质量值分布,只要大部分都高于20,那么就比较正常。
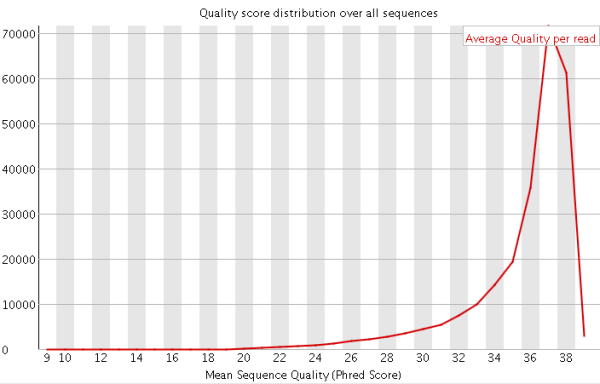
在第2节里面我也提到了关于Q20和Q30的比例是我们衡量测序质量的一个重要指标。这其实也是从这里来进行体现的,一般来说,对于二代测序,最好是达到Q20的碱基要在95%以上(最差不低于90%),Q30要求大于85%(最差也不要低于80%)。
2)read各个位置上碱基比例分布
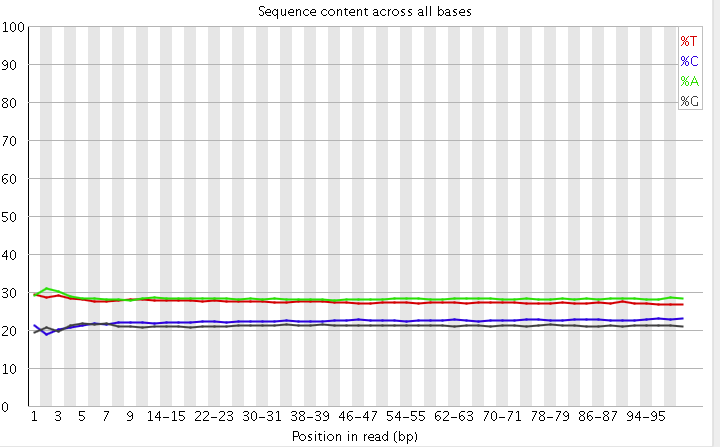
这个是为了分析碱基的分离程度。何为碱基分离?我们知道AT配对,CG配对,假如测序过程是比较随机的话(随机意味着好),那么在每个位置上A和T比例应该差不多,C和G的比例也应该差不多,如上图所示,两者之间即使有偏差也不应该太大,最好平均在1%以内,如果过高,除非有合理的原因,比如某些特定的捕获测序所致,否则都需要注意是不是测序过程有什么偏差。
3)GC含量分布图
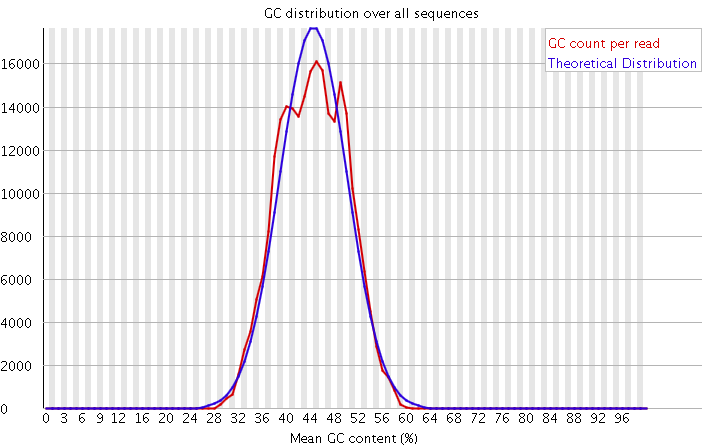
GC含量指的是G和C这两种碱基占总碱基的比例。二代测序平台或多或少都存在一定的测序偏向性,我们可以通过查看这个值来协助判断测序过程是否足够随机。对于人类来说,我们基因组的GC含量一般在40%左右。因此,如果发现GC含量的图谱明显偏离这个值那么说明测序过程存在较高的序列偏向性,结果就是基因组中某些特定区域被反复测序的几率高于平均水平,除了覆盖度会有偏离之后,将会影响下游的变异检测和CNV分析。
4)N含量分布图
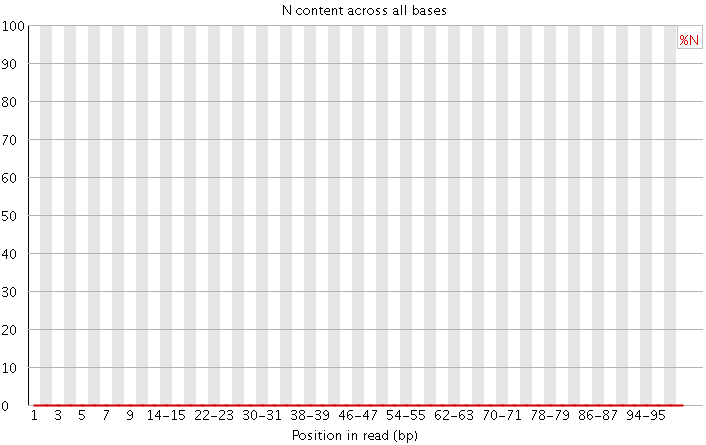
N在测序数据中一般是不应该出现的,如果出现则意味着,测序的光学信号无法被清晰分辨,如果这种情况多的话,往往意味着测序系统或者测序试剂的错误。
5)接头序列

在第1节 测序技术里面我们提到了在测序之前需要构建测序文库,测序接头就是在这个时候加上的,其目的一方面是为了能够结合到flowcell上,另一方面是当有多个样本同时测序的时候能够利用接头信息进行区分。当测序read的长度大于被测序的DNA片段【注】时,就会在read的末尾测到这些接头序列(如下图)。一般的WGS测序是不会测到这些接头序列的,因为构建WGS测序的文库序列(插入片段)都比较长,约几百bp,而read的测序长度都在100bp-150bp这个范围。不过在进行一些RNA测序的时候,由于它们的序列本来就比较短,很多只有几十bp长(特别是miRNA),那么就很容易会出现read测通的现象,这个时候就会在read的末尾测到这些接头序列。
【注】这些DNA片段也常被我们称之为“插入片段”
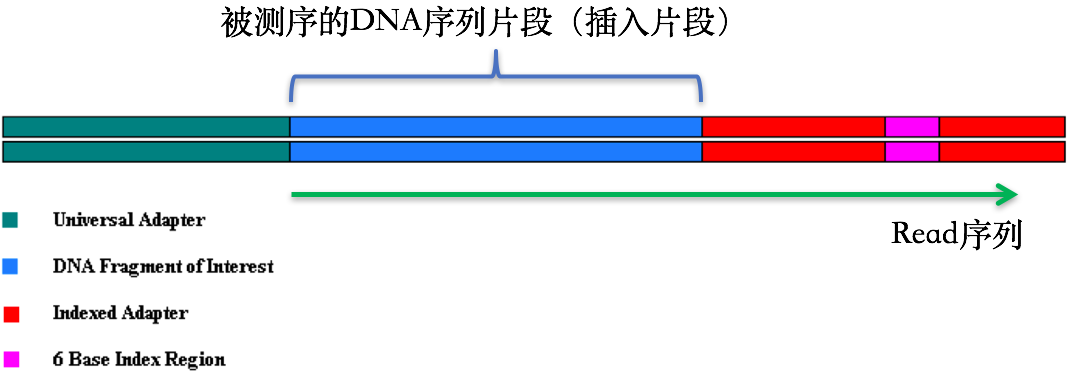
最后,这些被测到的接头序列和低质量碱基一样都是需要在正式分析之前进行切除的read片段。
当我们看完了上面的这些结果之后就可以比较清楚地了解一个测序数据的概况了。
那么,说了这么多,上述提到的FastQC该怎么用呢?
FastQC的安装非常简单,我们可以通过网页搜索或者直接到它的主页上下载最新的版本。
也可以在终端通过wget命令下载:
$ wget https://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.5.zip ./
解压之后,修改文件夹中fastqc的权限,就可以直接运行了:
$ unzip fastqc_v0.11.5.zip $ cd FastQC $ chmod 755 fastqc
FastQC的运行非常简单,直接在终端通过命令行是最有效直接的,下面我给出一个例子:
$ /path_to_fastqc/FastQC/fastqc untreated.fq -o fastqc_out_dir/命令比较简单,这里 唯一值得注意的地方就是 -o 参数用于指定FastQC报告的输出目录,这个目录需要事先创建好,如果不指定特定的目录,那么FastQC的结果会默认输出到文件untreated.fq的同一个目录下。它输出结果只有两个,一个html和一个.zip压缩包。
$ tree fastqc_out_dir/
我们可以直接通过浏览器打开html,就可以看到FastQC给出的所有结果,zip压缩包解压后,从中我们也可以在对应的目录下找到所有的QC图表和Summary数据。

除了上述用法之外,FastQC支持同时输入多个fq文件(或者以通配符的形式输入fq),当我们的fq文件比较多时,这种用法会比较方便,如:
$ /path_to_fastqc/FastQC/fastqc /path_to_fq/*.fq -o fastqc_out_dir/
这个链接是FastQC官网给出的一个Online报告的模板,方便参考。
切除测序接头序列和read的低质量序列
前面关于如何认识fq数据的事情已经说完了,接下来是我们本篇文章中最后的一个重点——去除测序接头和低质量序列!
当我们理解了fq数据之后,做这些过滤就不会很难,你也完全可以自己编写工具来进行个性化的过滤。目前也已有很多工具用来切除接头序列和低质量碱基,比如SOAPnuke、cutadapt、untrimmed等不下十个,但这其中比较方便好用的是Trimmomatic(也是一个java程序)、sickle和seqtk。Trimmomatic的好处在于,它不但可以用来切除illumina测序平台的接头序列,还可以去除由我们自己指定的特定接头序列,而且同时也能够过滤read末尾的低质量序列,sickle和seqtk只能去除低质量碱基。具体的原理就是通过滑动一定长度的窗口,计算窗口内的碱基平均质量,如果过低,就直接 往后全部切除,注!意!不是挖掉read中的这部分低质量序列,而是像切菜一样,直接从低质量区域开始把这条read后面的所有其它碱基全!部!剁!掉!否则就是在人为改变实际的基因组序列情况。
如果下机的fq数据中不含有这些测序接头,那么我们除了trimmomatic之外,也可以直接使用sickle(同时支持PE和SE数据)或者seqtk(仅支持SE),这两个处理起来会更快,消耗的计算资源也更少。
现在我们说回如何用Trimmomatic构造序列过滤流程。
首先是安装Trimmomatic。我们可以到它的官网上获取最新的版本,下载打包好的binary即可,如果打算看它具体的代码,可以在github上找到。

下载后,直接解压,目录下的trimmomatic-*.jar(我下载的是0.36版本)就是执行程序,可以直接使用java来运行。
$ java -jar trimmomatic-0.36.jar
同个目录下还有一个名为adapters的文件夹,这个文件夹中的内容对于我们去除接头序列来说非常重要。其中默认存放的是illumina测序平台的接头序列(fasta格式),在实际的使用过程中,如果需要去除接头,我们需要明确指定对应的序列作为输入参数。
那么这些接头序列具体该如何选择呢?一般来说,目前的HiSeq系列和MiSeq系列用的都是TruSeq3,TruSeq2是以前GA2系列的测序仪所用的,已经很少见了。这些信息都可以在illumina的官网上找到,至于具体该用PE(Pair End)还是SE(Single End)就按照具体的测序类型进行选择就ok了。如果用的不是illumina测序平台,那么我们也可以按照adapters文件夹下的这些文件的格式做一个新的接头序列,然后再作为参数传入。不过在自定义接头序列的时候,命名时有一些小的细节需要注意,可以参考Trimmomatic的主页文档(The Adapter Fasta),这里就不展开了。
Trimmomatic有两种运行模式:PE和SE。顾名思义,PE就是对应Pair End测序的,SE则是对应Single End测序的。
$ java -jar trimmomatic-0.36.jar Usage: PE [-version] [-threads <threads>] [-phred33|-phred64] [-trimlog <trimLogFile>] [-quiet] [-validatePairs] [-basein <inputBase> | <inputFile1> <inputFile2>] [-baseout <outputBase> | <outputFile1P> <outputFile1U> <outputFile2P> <outputFile2U>] <trimmer1>... or: SE [-version] [-threads <threads>] [-phred33|-phred64] [-trimlog <trimLogFile>] [-quiet] <inputFile> <outputFile> <trimmer1>... or: -version
下面我分别给出例子来进行说明:
PE模式,HiSeq PE测序:
$ java -jar /path/Trimmomatic/trimmomatic-0.36.jar PE -phred33 -trimlog logfile reads_1.fq.gz reads_2.fq.gz out.read_1.fq.gz out.trim.read_1.fq.gz out.read_2.fq.gz out.trim.read_2.fq.gz ILLUMINACLIP:/path/Trimmomatic/adapters/TruSeq3-PE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:5 TRAILING:5 MINLEN:50
SE模式,HiSeq SE测序:
$ java -jar /path/Trimmomatic/trimmomatic-0.36.jar SE -phred33 -trimlog se.logfile raw_data/untreated.fq out.untreated.fq.gz ILLUMINACLIP:/path/Trimmomatic/adapters/TruSeq3-SE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:5 TRAILING:5 MINLEN:50
我们可以看到PE和SE,顾名思义,分别代表了 ‘PE模式’和‘SE’模式。
同时需要明确指明质量值体系是Phred33还是Phred64,默认是Phred64,这需要特别注意,因为我们现在的测序数据基本都是Phred33的了,所以一定要指定这个参数。
剩下的就是输入的fq和输出的fq,可以用-basein和-baseout指定,也可以不用(如上例子),以及被过滤掉的fq要输出到文件。细心的读者可能已经发现这里PE和SE有一个区别:
在SE模式中,是不需要指定文件来存放被过滤掉的read信息的,后面直接就接Trimmer信息!这是需要注意到的一个地方。
关于后面的Trimmer信息,规定了很多切除接头序列和低质量序列的细节,我挑重点的说,具体如下:
- ILLUMINACLIP,接头序列切除参数。LLUMINACLIP:TruSeq3-PE.fa:2:30:10(省掉了路径)意思分别是:TruSeq3-PE.fa是接头序列,2是比对时接头序列时所允许的最大错配数;30指的是要求PE的两条read同时和PE的adapter序列比对,匹配度加起来超30%,那么就认为这对PE的read含有adapter,并在对应的位置需要进行切除【注】。10和前面的30不同,它指的是,我就什么也不管,反正只要这条read的某部分和adpater序列有超过10%的匹配率,那么就代表含有adapter了,需要进行去除;
【注】测序的时候一般只会测到一部分的adapter,因此read和adaper对比的时候肯定是不需要要求百分百匹配率的,上述30%和10%其实是比较推荐的值。
SLIDINGWINDOW,滑动窗口长度的参数,SLIDINGWINDOW:5:20代表窗口长度为5,窗口中的平均质量值至少为20,否则会开始切除;
- LEADING,规定read开头的碱基是否要被切除的质量阈值;
- TRAILING,规定read末尾的碱基是否要被切除的质量阈值;
- MINLEN,规定read被切除后至少需要保留的长度,如果低于该长度,会被丢掉。
此外,另一个值得注意的地方是,Trimmomatic的报错给出的提示信息都比较难以定位错误问题(如下图),但这往往都只是参数用没设置正确所致。
Exception in thread "main" java.lang.RuntimeException: Unknown trimmer: u.trim.txt at org.usadellab.trimmomatic.trim.TrimmerFactory.makeTrimmer(TrimmerFactory.java:70) at org.usadellab.trimmomatic.Trimmomatic.createTrimmers(Trimmomatic.java:59) at org.usadellab.trimmomatic.TrimmomaticSE.run(TrimmomaticSE.java:303) at org.usadellab.trimmomatic.Trimmomatic.main(Trimmomatic.java:85)
小结
数据质控的内容终于都讲完了,接下来第4节就是主流程的构建。
1F
PE数据用trimmomatic进行质控会生成四个文件,除了两个paired 文件还有两个unpaired文件,unpaired文件在后续分析中还需要用到它们吗?该怎么用?